


نمایش آذرخش در لوله آزمایش
شهریور ۳۱, ۱۳۹۷
مقدمهای بر لومینسانس
مهر ۸, ۱۳۹۷
به نام خدا و با سلام خدمت دوستان عزیز به خصوص دانشجویان عزیز در قسمت قبل من نرم افزار مستر نوا براتون توضیح دادم اما امروز میخواهم جزئیات بیشتری از این برنامه بگویم .
نرم افزار بسیار قوی و پرکاربرد Mestrec را که مخصوص کار بر روی طیف های NMR هست رو تا جایی که وقت اجازه می ده آموزش بدم. نسخه portable و بدون نیاز به نصب این نرم افزار که از ظاهر و کیفیت بسیار بهتری نسبت به نسخه نصبی برخورداره نامش mestrenova هست که من برای آموزش در نظر گرفتم. مسلما این نرم افزار منوهای زیادی داره که خیلی از آنها ممکن است به کار نیان اما چند تا از آنها هستند که برای کار بر روی طیف های NMR ضروری هستند.
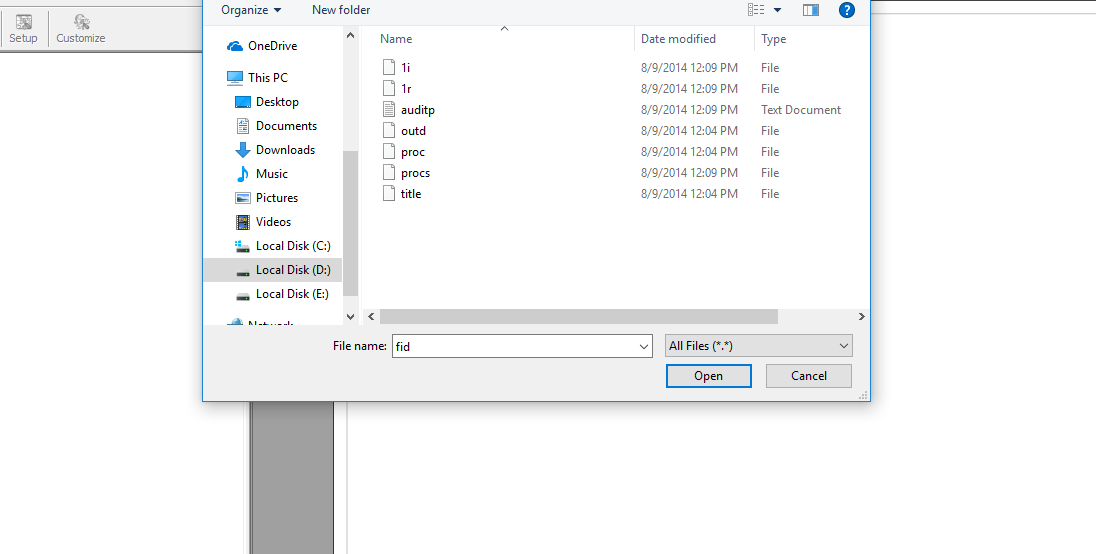
این نرم افزار مثل هر نرم افزار دیگه ای دارای منوهایی هست. اولین کاری که باید بکنیم این هست که داده های NMR را وارد این نرم افزار بکنیم. فقط کافیه است که بعد از باز کردن نرم افزار فایل FID رو به درون صفحه محیط کار drag کنیم.
تمام dataهای NMR در فایلی به نام FIDذخیره می شود که میتوان طیفهای رادر mnova مشاهده کرد.

درون پوشه ی Pdata :1i ,1r هم وجود دارد که میتواند کار فایل FIDرا انجام دهد که از اینها زمانی استفاده میکنیم که به هر دلیلی فایل FID پاک شده است ابتدا سراغ 1rرفته بعد 1iکه اطلاعات در این پوشه به صورت فوریه ترنسفر هسته یعنی تعداد سیگنالها یا پالسهایی که زده شده ممی باشد به تبدیل انهای گزینه F1(orginal FID) می زنیم .
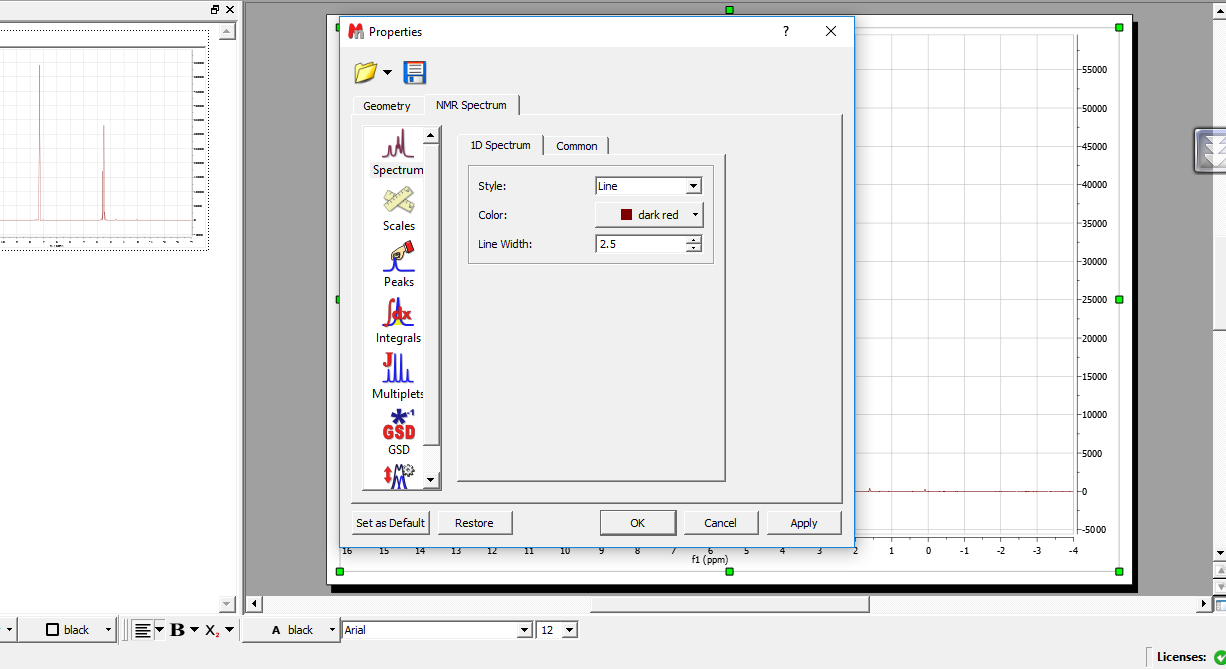
حذف چهار خونههای پس زمینه:
از سه طریق میتوان منوی propertis باز کرد :
1-double click در صفحه ی طیف
2- کلیک راست در صفحه
3- در نوار ابزار گزینه ی edit
پس از باز کردن پنجره properties تب scail ← Grid حذف تیک ها
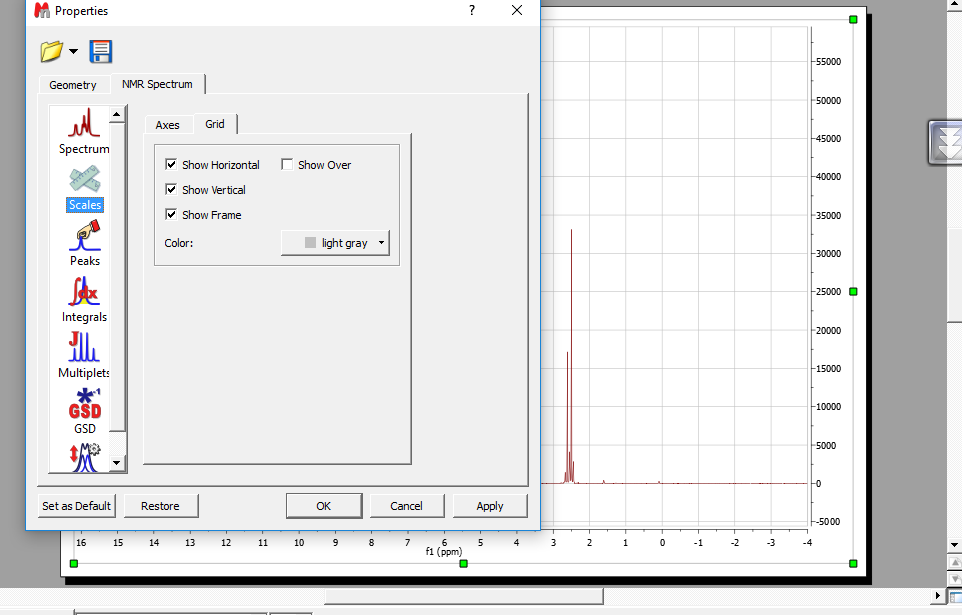
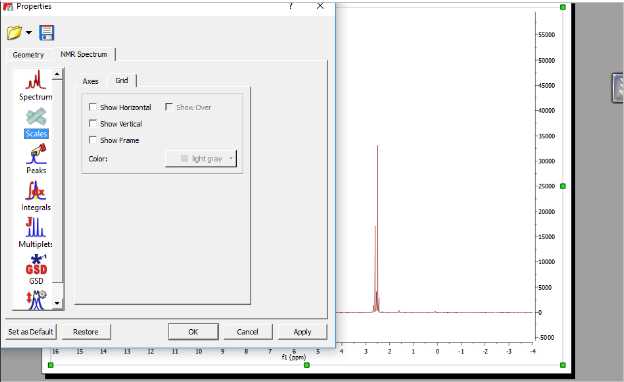
تغییر رنگ خطوط چهارخونه
Properties → scal → Grid → color
حذف title :
برای حذف title روی طیف NMRکه توسط مسئول NMRنوشته شده (sample code ) کلیک راست properties سپس spectrum سپس commonو بعد تیک گزینه ی title برمی داریم.
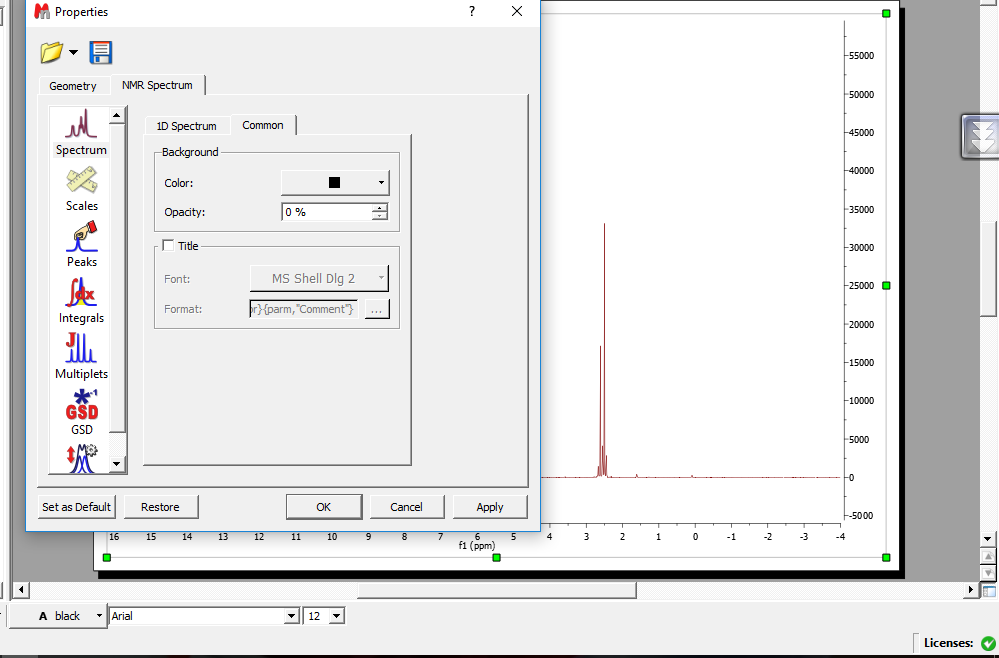
اگر بخواهیم در حافظه برنامه بماند که هیچ گاه بر روی طیف ها title نباشد گزینهی set as default را می زنیم .
تغییر رنگ در style طیف ها
انتخاب اینکه طیف ها حطی باشد یا نقطه نقطه ابتدا کلیک راست سپس properties وبعد ID spectrum و درنهایت style را میزنیم
نکته برای پایان نامه طیفها باید خطی سیاه و سفید و باسایز ۴ باشد .
تغییر رنگ
کلیک راست وبعد properties بعد ID spectrum در نهایت color
تغییر ضخامت
کلیک راست وبعد properties بعد ID spectrum در نهایت line width
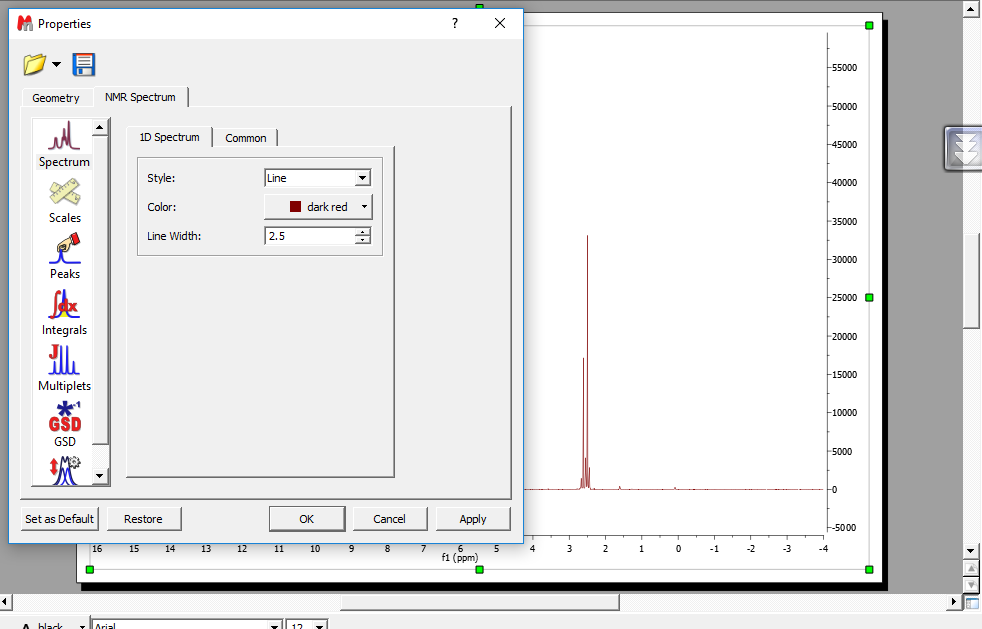
گزینه بعدی scales هست که در tab به نام Axes انواع تنظیمات مربوط به محور افقی یا ppm و محور عمودی یا intensity که معمولا در NMR حذف میشه رو انجام داد. رنگ , فونت و margin رو برای هردو محور میشه تغییر داد. برای محور افقی یا PPM میشه برچسب و واحد و رقم بعد از اعشار رو تعیین کرد و همینطور برای محور عمودی یا vertical میشه برچسب یا رقم بعد از اعشارش رو تعیین کرد. همینطور در tab دیگر به نام Grid ما می تونیم یک شبکه در پشت طیف با تنظیماتی که می خواهیم قرار بدیم ولی خوب معمولا این شبکه برداشته میشود.
گزینه بعدی integrals هست که در اون در قسمت label می تونید برچسب و فونت و رنگ و رقم بعد از اعشار و موقعیت پیک رو تعیین کنید و در قسمت curve تنظیمات مربوط به منحنی های انتگرالی روی پیک ها رو انجام بدید.
نکته وقتی نمونه داخل دستگاه قرارمی گیرد و بافشار هوا پایین می رود صدای تق میدهد که در این زمان مسئول دستور LOCK را میدهد یعنی طیف NMRروی هسته LOCKمیکند که هسته هم D است و حلال مورد استفاده دوتریم است وقتی LOCK را می زند انواع حلال ها رانوشته مانند CDCL3 و CD3 CN که اولین کاری که مسئول میکند LOCK کردن نمونه است.
تشخیص LOCK دستگاه
در طیف هایی که داریم اگر ندانیم چه حلالی استفاده شده
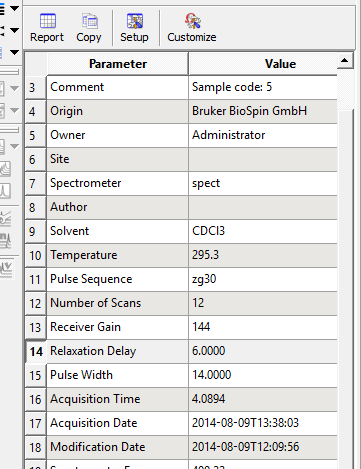
پنجره ای که دارای تمامی مشخصات طیف ابتدا نوار ابزار گزینه ی VIEW سپس TABLE وبعد PARAMETRES شامل

موارد مورد نیاز در PARAMETRES شامل دما و نام حلال و زمان و N.s و نوع هسته S.W فرکانس دستگاه
نکته :N.S تعداد پالس های زده شده است که مثلا N.s اگر ده بود اگر طیفمان خوب نبود می توانیم تعداد پالس ها را 50 کنیم.
SPECTRAL WIDTH (S.W)اندازه طیف را نشان می دهد که اگر تقسیم بر ۴۰۰ قدرت دستگاه کنیم بازه ی طیف برحسب PPMحاصل میشود.
Spectrometry frequency قدرت دستگاه نشان میدهد
Relaxation Delay مدت زمانی که زده شدن هر پالس طول میکشد مثلا ۴ ثانیه که به ان D1 هم گفته میشود پالس زده میشود ماده برانگیخته میشود پایین می اید که این پدیده قبل و بعد از زده شدن هم 1ثانیه طول میکشد هرپالس ۶ ثانیه طول میکشد
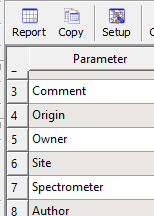
اگر بخواهیم داده های مورد نظر در صفحه ی parameter باقی می ماند
در پنجره ی parameter در بالای پنجره customizeسپس فقط مواردی که میخواهیم تیک میزنیم

اگر بخواهیم اطلاعات در کنار طیف درج شود.
در پنجره parameter گزینه ی report
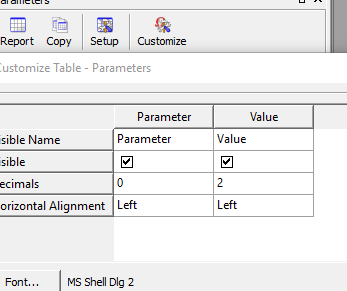
اگر بخواهیم font ,color و………… را تنظیم کنیم
در پنجره parameter گزینه ی setup
نکته از روی مقاله ی مرجع ( organometalics 2010.29.2176-2179.)میتوان محل ظاهرشدن پیک های حلال را پیدا کرد که برحسب ppmمی باشد مثلا استون در 2.05ppmظاهر میشود پس وقتی دستگاه را lockمیشود به طورهوشمندانه محل ظاهرشدن پیک حلال را میداند.
وقتی بخواهیم محل طیف را به طول دقیق بخوانیم TMS را انتخاب میکنیم و روی محل پیک مورد نظر گذاشته وعدد ظاهر میشود .
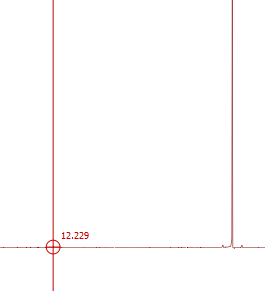
نکته : کار بعدی تعیین کردن chemical shift ها هست که برای این کار شما ابتدا باید پیک مربوط به حلال رو lock کنید. در این نمونه ای که من برای شما نشان میدم حلال CDCl3 هست که ناخالصی CHCl3 باید دقیقا در 7.26 قرار بگیره. برای این کار در نوار ابزار بالا روی icon با شکل TMS و به نام reference کلیک می کنید. خط قرمزی روی صفحه ایجاد می شه که اون رو روی پیکی که می دونید مربوط به حلالتون هست می برید و کلیک می کنید که بلافاصله پنجره ای به نام Reference analog f1 باز می شه که می تونید در قسمت New shift جابجایی شیمیایی مربوط به حلالتون رو وارد کنید.
نکته اگر گزینه ای در منونمایان نبود (منظور نوار ابزار بالای صفحه )
هر کدام از منوهای موردنظر را اضافه میکنیم
راست کلیک سپس CUSTOMIZE TOOLBAR
نشان دادن طیف به صورت کلی
یکی از مهم ترین کارهایی که میشودانجام داد : کل طیف یا یک محدوده خاصی از طیف رو بزرگ یا کوچک کنیم برای این کار کافیه که روی icon به شکل ذره بین با علامت مثبت یا منفی کلیک کنیم که به ترتیب Zoom in و Zoom out نام دارند . اگر روی zoom in کلیک کردید باید اون منطقه از طیف رو که می خواهید expand کنید با کلیک موس و نکه داشتن کلیک انتخاب کنید اما برای zoom out فقط باید روی یک نقطه سفید از طیف کلیک کنید. اگر خواستیم که به یک باره کل طیف رو ببینیم می تونیم از icon کنار zoom out به نام full spectrum استفاده کنیم.
![]()
اولین از سمت چپ برای ZOOM بزرگ کردن
دومی ZOOM کوچک کردن
سومی کل پیک را نشان میدهد
این هست که با Icon که به شکل دست هست می تونید کل طیف رو بالا و پایین ببرید. برای این کار کافیه که روی icon کلیک کنید و بعد نشانگر موس رو روی صفحه طیف ببرید و کلیک کنید و نگه دارید و طیف رو بالا و پایین ببرید.
![]()
پیدا کردن محل SIGNAL حلال ها
در میان حلال های دوتره هر چقدر هم خالص باشد مثلا CDCl3 اما باز هم میتواند CHCl3 هم داشته باشد
اگر ظاهر شدن پیک حلال با عددی که در مقاله مرجع داده شده اختلاف داشته باشد برای جبران ان گزینه ی TMSرا استفاده میکنیم
کلیک روی گزینه ی TMS سپس بردن نشانه روی پیک حلال وبعد کلیک روی پیک ودر نهایت بازشدن پنجره در NEWSHIFT و وارد کردن عدد مرجع در مقاله در نهایت تمام طیف اینگونه SHIFT میکنند .
اگر ندانیم دقیقا پیک حلال در کجا می افتد :
TMSسپس بردن نشانه روی پیک وبعد کلیک روی پیک وسپس باز شدن پنجره و سپس SOLVENT پنجره ای باز میشود که شامل همه ی حلال ها و SHIFT شان باز میشود وحلال را اتنخاب میکنیم .
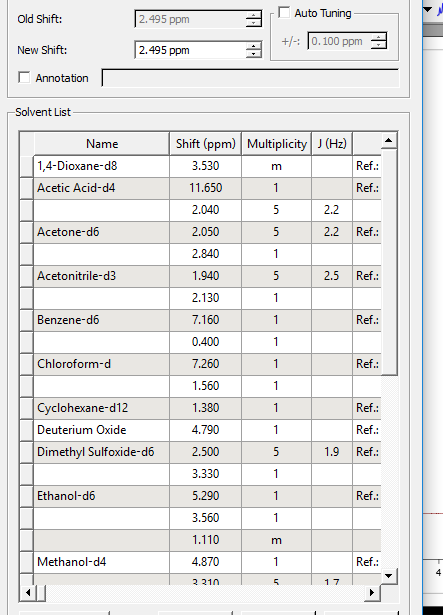
در پنجره ای که شامل حلال ها در زیر حلال سیگنال هایی نوشته شده که مربوط به اب است که این نشان میدهد تمام این حلال ها اب وجود دارد .
گزینه ی PEAK PIKING
تعیین جایجایی شیمیایی
کار بعدی تعیین جابجایی های شیمیایی مربوط به دیگر پیک هاست که نسبت به حلال قرار دارند. برای این کار روی icon با نام peak picking که در سمت راست TMS قرار داره کلیک می کنید که یک drop menu باز می شه. اولین گزینه Automatic هست که وقتی روی اون کلیک می کنید جابجایی شیمیایی تمام پیک ها و حتی ریزترین پیک ها رو روی پیک مربوطه قرار میده. دومین گزینه options هست که می تونید تغییرات مد نظر خودتون رو در پنجره ای که باز میشه و نامش peak peaking options هست اعمال کنید (البته تا به حال این گزینه به کار من نیامده). سومین گزینه manual threshold هست که در اون می تونید یک قسمت خاص از طیف رو که نیاز دارید جابجایی های شیمیاییش رو تعیین کنید. چهارمین peak by peak هست که در اون می تونید پیک ها رو دونه به دونه و فقط اونهایی رو که نیاز دارید جابجایی های شیمیاییش رو تعیین کنید. گزینه های report و copy مهم نیستند. گزینه delete manually جابجایی های شیمیایی اون قسمت خاصی رو که مد نظرتون هست رو پاک می کنه و delete all همه رو پاک می کند .
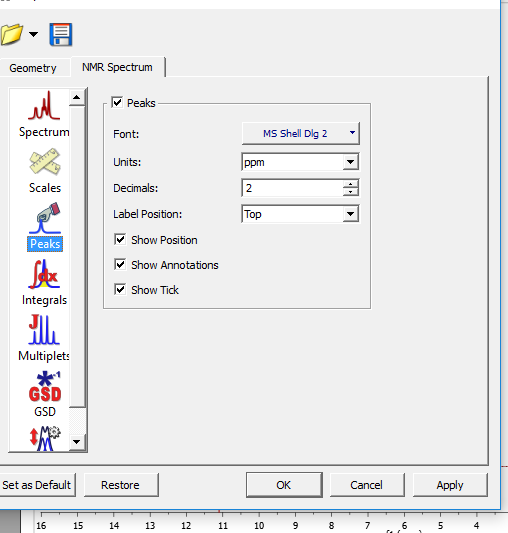
در این طیف ها جابجایی که سیستم فکرمیکند ما پیک زده ایم ک خط کوچک بالای پیک می زند برای برداشتن این خط کوچک
ابتدا PROPERTIES سپس PEAKS وبعد تیک SHOW TICK برداشتن
معمولا پیک حلال را فقط وسطی اش را میزند .
خودش یک قابلیت دارد که سیگماها را طبق ژورنالی که میخواهیم در ان گزارش کنیم برای ما وارد میکند که یکسری از اینها را باید حذف کرد چون برای ما فقط پیکهای اصلی مهم هستند .
در نوار ابزار VIEW سپس TABLEوبعد PEAKS پنجره ای باز میشودکه مثلاHNMR را می اوردکه شیفت هایی که شیمیایی را هم می اورد که بیشتربرای طیفهای کربن خوب است اما ماباید در پیکهای اصلی را تشخیص داده وبدانیم که کدام باید REPORTشود وکدام سیگنال بیشتر به ترکیبات آلی میخورد.
در پنجره ی PEAKS سپس REPORT SPECIAL درنهایت SETUPدر آخر انتخاب یکی از ژورنال های در واقع TYPE زورنال انتخاب میکنیم کپی در ورد میکنیم .
اما بازدن گزینه REPORT در بالا فقط در طیف NMR ظاهر میشود.
هنگام کپی کردن اگر CTRL+Cرا بزنیم طیف راکپی میکنیم .
حتما حتما برای کپی کردن DATAها باد انتخاب کنیم و SELECT ALLرا بزنیم وکپی PASTرا در ورد بزنیم .در PEAK PICKING اگر SHOW PEAKرا بدون تیک بزنیم CHEMICAL SHIFTها ظاهر نمیشود اگر دوباره تیکش را بزنیم دوباره به وجود می اید.
کلیک راست PROPERTIES سپس PEAKوبعد تیک را برداشته وسیگما همگی مخفی شده و همان نقش SHOW PEAK را دارد.
کار مهم بعدی تعیین شکافتگی یا J هست که در این نرم افزار متاسفانه ما نمی تونیم مقدار J رو روی صفحه ثبت کنیم و فقط می تونیم اون رو اندازه بگیریم. برای این کار روی icon به شکل علامت مثبت بزرگ آبی رنگ در نوار ابزار بالای صفحه به نام Crosshair کلیک می کنیم. بعد از کلیک دو خط آبی رنگ عمود بر هم ظاهر می شن که نقطه طلاقی این دو خط رو روی نقطه ابتدایی شکافتگی می بریم و کلیک می کنیم و کلیک رو نگه می داریم و به انتهای قسمت شکافتگی می بریم. به محض کلیک کردن دو خط قرمز عمود بر هم نیز ظاهر می شود و همچنین یک پنجره به نام info view که در پایین این پنجره عدد قدر مطلق B-A بر حسب هرتز میزان شکافتگی مورد نظر شما رو نشون می دهد. مسلما برای اندازه گیری دقیق میزان J شما باید اون منطقه از طیف رو تا جایی که میشه expand یا zoom in کنید.

یکی از مهمترین کارهایی که با این نرم افزار میشه کرد تعیین انتگرال پیک هاست مخصوصا در مورد NMR هسته هیدروژن. برای این کار ابتدا icon با نام integration رو که با شکل یک علامت انتگرال و یک پیک در کنارش دیده میشه رو از نوار ابزار بالا انتخاب می کنیم. ممکن هست به خاطر کمبود جا در نوار ابزار بالا این icon دیده نشه اما یک icon از نوع drop هست که اگر روی آن کلیک کنیم بقیه icon ها رو نشون میده و معمولا icon انتگرال در بین اونهاست. وقتی که روی این icon کلیک کنید یک menu باز میشه که گزینه اول Autodetect regions هست و همه پیک ها رو با هم انتگرال میده که معمولا به کار نمیاد. گزینه بعدی predefined regions هست که در این نرم افزار خاموش هست. گزینه بعدی options هست که وقتی روی آن کلیک می کنید پنجره Auto-integration options هست که می تونید options رو تغییر بدید. گزینه بعدی manual هست که گزینه اصلی به حساب میاد. وقتی روی این گزینه کلیک می کنید icon موس به شکل علامت انتگرال در میاد. برای کار ابتدا باید روی یک پیک که تعداد هیدروژن هاش یک باشه یا اینکه تعداد رو خودتون می دونید کلیک کنید و با نگه داشتن کلیک در کل پیک اون رو انتخاب کنید که نرم افزار به صورت default عدد یک رو برای اولین پیک نشون میده. بعد بقیه پیک ها رو انتخاب می کنید که نسبت به انتگرال پیک اولیه انتگرال اونها نمایش داده میشه. گزینه های بعد delete manually و delete all هم به ترتیب برای پاک کردن پیک خاص و کل پیک ها هست.
یکی از کار های جالبی که میشه با این نرم افزار انجام داد وارد کردن inset یا بزرگ کردن یک قسمت از پیک و نشون دادنش داخل همون پیک هست. برای این کار icon به نام expansion رو در که در سمت راست icon به شکل دست قرار داره رو روش کلیک می کنید و اون قسمت از طیف رو که می خواید inset مربوط بهش رو در طیف داشته باشید رو با کلیک و نگه داشتن کلیک روی اون پیک انتخاب می کنید. نکته جالب اینجاست که تمام کارهایی که با طیف اصلی می تونید انجام بدید رو با inset هم می تونید انجام بدید مثل بزرگ کردن بیشتر یا قرار دادن جابجایی شیمیایی. حتی می تونید مستطیل inset رو بزرگ یا کوچک هم بکنید. برای این کارها باید inset رو انتخاب کنید که بعد از انتخاب مربع های سبز دور مستطیل inset ظاهر میشه. حتی می تونید بیشتر از یک inset روی طیفتون داشته باشید. همچنین می تونید inset رو روی صفحه طیف با فلش های جهت روی کی بورد جابجا کنید. اگر خواستید inset رو پاک کنید از کلید delete روی کی بورد استفاده کنید
![]()
یکی دیگه از icon های مفید و قابل استفاده در نوار ابزار بالا icon به نام cut هست که به این کار میاد که اگر یک قسمت از طیف هیچ پیکی نداره و شما می خواید بدون دست زدن به بقیه طیف اون قسمت رو حذف کنید باید از این icon استفاده کنید. برای این کار icon به شکل قیچی رو در نوار ابزار بالا که در سمت راست icon مثبت آبی بزرگ (که برای اندازه گیری J بود) رو انتخاب کنید و اون قسمت از طیف رو که می خواید حذف شه رو انتخاب کنید که بلافاصله حذف می شه و یک علامت رو به شکل // در محور X ها که مربوط به جابجایی شیمیایی هست قرار میده که این علامت نشون میده که بین این دو علامت اسلش حذف شده. در drop menu کنار این icon یک گزینه به نام auto cut هست که همه قسمت های بدون پیک طیف رو حذف می کنه. بقیه گزینه ها هم مشخص هستند
از نوار ابزار بالا دو icon دیگر به نام های baseline correction و automatic phase correction هستند که برای صاف کردن baseline و درست کردن پیک هایی که به سمت پایین رفتند مناسب است. یک نکته اینکه در drop menu در automatic phase correction یک گزینه به نام magnitude هست که می تونید از اون هم برای بهتر کردن طیف استفاده کنید.
یک نکته که شاید همه بدونن این هست که در منو فایل برای save کردن طیف بعد از اعمال تغییرات به انواع فرمت های مختلف هست. اگر طیف رو به همین شکلی که آماده کردید می خواید save کنید به طوری که با خود نرم افزار باز بشه و دوباره روش کار کنید باید با پسوند mnova ذخیره کنید و بعدا دوباره فایل mnova مربوطه رو به داخل نرم افزار بکشید. گزینه export to pdf هم از طیفتون pdf می سازد.
همونطور که تا به حال دیدید در قسمت سمت چپ محیط نرم افزار یک قسمت به نام pages هست که در اونجا می تونید هر چند تا طیفی رو که وارد نرم افزار می کنید رو به صورت کوچک شده ببینید که روی هر کدوم کلیک کنید اون طیف رو نشون میدهد
از همین قسمت pages میشه طیف ها رو برای مقایسه روی هم گذاشت. دو یا سه یا هر چند تا طیف که می خواید رو می تونید برای مقایسه وبررسی شیفت یک پیک خاص در طیف وارد کنید. برای این کار کلید control رو نگه دارید و طیف ها رو انتخاب کنید. به محض این کار در سمت چپ دو icon که تا به حال خاموش بودند روشن می شوند که به نام های stack selected spectra و superimpose selected spectra که طیف ها رو برای مقایسه در دو حالت مختلف روی هم می گذارند. و همینطور icon های پایین تر که از اونها برای بالا و پایین بردن طیف ها و عوض کردن ترتیب اونها و همینطور بلند یا کوتاه کردن یکی از طیف ها استفاده کردو
گاهی اوقات بعضی طیف ها دارای baseline های پهن هستند به طوری که پیک های اصلی به درستی دیده نمی شن. برای همین باید مقداری smooth بشن. برای همین منظور باید از منوی processing در بالا گزینه smoothing رو انتخاب کنیم که پنجره ای به نام smoothing along f1 باز میشه. در قسمتmethod گزینه Savitzky-Golay وجود داره که در قسمت mode می تونید اون رو روی high بزارید که تا اندازه زیادی طیف بهتر میشه و همینطور می تونید method رو به moving average filter تغییر بدید و عدد مقابل کلمه span رو بیشتر کنید البته تا جایی که به شکل پیک ها آسیب نزنه. مثلا اینکه دابلت رو سینگلت نکنه در قسمت method دو گزینه دیگر به نام های whittaker smoother و wavelets وجود دارند که به اندازه دو تای دیگه پرکاربرد نیستند ولی می تونید از اونها هم استفاده کنید.
اما در انتها یک قسمت خیلی به درد بخور رو می خوام بگم که شما در این قسمت می تونید ظاهر طیفتون رو بعد از تغییراتی که در بالا اشاره شد هر طور که خواستید تغییر بدید. اگر روی صفحه اصلی طیف right click کنید یک منو باز میشه که اگر روی قسمت properties کلیک کنید یک صفحه باز میشه به نام properties. در قسمت سمت چپ این پنجره در واقع یک ستون هست که در این ستون گزینه های spectrum, scales, peaks, Integrals و …..دیده میشود.
نکته آخر راجع به این قسمت این هست که در بالای پنجره properties یک icon مربوط به save هست که می تونید تمام تغییراتی رو که داید برای همیشه save کنید و همیشه طیفتون با این تنظیمات دیده بشه. در پایین پنجره Properties یک icon به نام set as default هست که تمام تنظیمات نرم افزاری رو مثل اول می کنه. Restore هم که در کنار اون هست برای برگردوندن تنظیماتی هست که در اون لحظه ایجاد کردید و هنوز save نشدن.
قبل از اتمام بحث, برای قرار دادن ساختار مولکول روی طیف برای مثلا پایان نامه میتونید از این نرم افزار استفاده کنید اما کیفیت خروجی خوب نمیشه برای همین ترجیحا برای این کار از نرم افزار های دیگه مثل پاورپوینت میشه استفاده کرد که کیفیت خیلی خوبی دارد.
فکرمی کنم تمام چیزهایی که لازم بود برای کار با طیف آموزش بدم رو ارائه کردم. امیدوارم که تونسته باشم تا حد خوبی این نرم افزار رو آموزش داده باشم.
مشاهده ویدیو های آموزشی
به امید روزهای بهتر وروشن تر
مائده کوهی
دانشجوی دکتری شیمی معدنی

رزومه انگلیسی



7 Comments
سلام
دست شما درد نکنه
عالی بود
سلام
درباره نمودارهای جی سی توضیح ندارید؟
سلام، ببخشید من وقتی فایل fid را باز می کنم، میزنه Unknown format of file”acqu” میزنه میشه راهنمایی کنید
سلام، ببخشید من وقتی فایل fid را باز می کنم، میزنه Unknown format of file”acqu” میزنه میشه راهنمایی کنید
p30download.ir/fa/entry/71365/
ُسلام تک تک مراحل رو از لینک بالا انجام بدید و نصب کنید در صورت خطا آنتی ویروس رو غیر فعال کنید.موفق ومانا باشید
سلام
بسیار عالی بود
بسیار واضح و قابل فهم
توضیحات جامع و کامل بود
سپاسگزارم
سلام چطوری میتونیم عددهای مربوط به انتگرال را زیر خط افقی منتقل کنیم؟